【NGS 次世代基因體資料科學】 基礎教學 03 使用fasterq-dump取得已發表的實驗的原始資料

一般已經發表的生物醫學領域的論文,如果有牽涉到定序都會把原始數據上傳。這裡介紹如何把它們用SRA Tools把資料撈回來,讓我們能夠重複一些重要的實驗結果。
本篇以NCBI的GEO為例,示範使用NCBI 的Gene Expression Omnibus (GEO) database 。假設我們從論文上的Method或Supplementary 得知GEO的網址為:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1010219
最下方有一個SRA Run :

點下去後會跳轉到SRA Run Selector的網站:
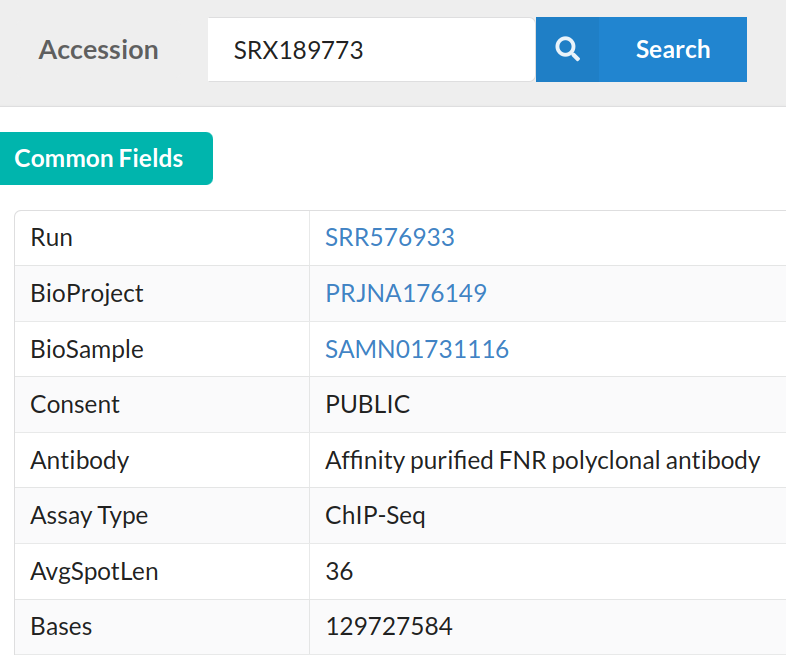
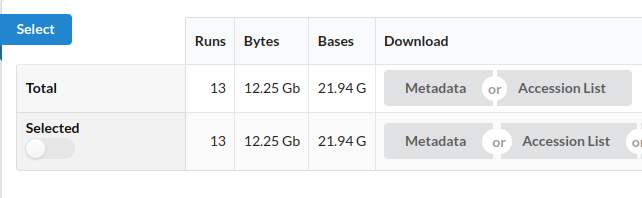
以這邊為例,可以拿到SRR開頭的run ID SRR576933 ,也有可能有多個run,可以點Assesion List一次取:
接著用fastq-dump或是fasterq-dump可以把對應的fastq檔下載回來,這裡示範使用fasterq-dump來把檔案下載回來
get_fastq.sh :
ShellScript
#!/bin/bash
# using fasterq-dump to download fastq
tmp_path='./tmp_'$1 # tmp path name , change if you want
thread_num=24 # your thread number
SRRID=$1 # SRR ID pass from shell
mkdir $tmp_path
export TMPDIR=$tmp_path
fasterq-dump "$SRRID" --split-3 \
--threads $thread_num --temp $tmp_path \
--progress
rm -r $tmp_path然後在終端機執行:
./get_fastq.sh SRR576933執行完成後會得到輸出
join :|-------------------------------------------------- 100%
concat :|-------------------------------------------------- 100%
spots read : 3,603,544
reads read : 3,603,544
reads written : 3,603,544就可以拿到實驗的fastq檔的raw data了
有了fastq檔之後,接下來通常就是拿去做sequence alignment,來進行後續的分析。
專案網址
是一個對 “下一個世代” 的醫療科技充滿熱血的 Bioinformatican
Related Posts

【NGS 次世代基因體資料科學】Gene2vec基因的分散式表徵
這裡紀錄如何利用論文Gene2vec提供的程式,並搭配自己的資料重新訓練模型,訓練基因的分散式表徵

【NGS 次世代基因體資料科學】使用bioinfokit繪製火山圖Volcano Plot
這裡介紹如何用現成的...
【NGS 次世代基因體資料科學】t-SNE簡介
何謂 t-SNE t...